

Researchers at the Swiss Federal Institute of Technology Lausanne (EPFL) have developed a new evaluation tool called Systema to test the accuracy of artificial intelligence models in predicting the effects of gene perturbations. The study, led by the MLBio laboratory (Biomedical Machine Learning Lab) at EPFL, was published in the journal Nature Biotechnology.
Gene perturbation refers to the artificial alteration of genes to observe their impact on cellular function. This approach is of great significance for understanding gene regulatory mechanisms and developing new therapies. With the growth in the scale of experimental data, artificial intelligence models are widely used to predict the effects of gene combinations that have not been experimentally tested. However, the reliability of their predictions still requires systematic evaluation.
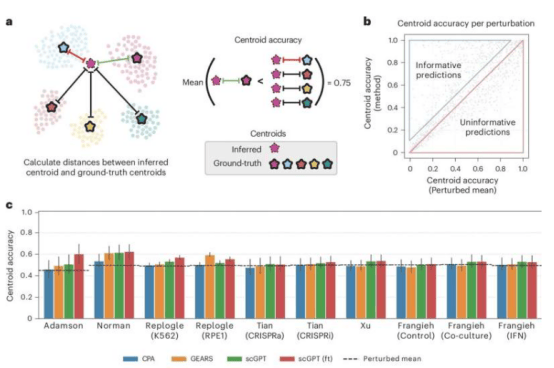
The research team compared the performance of current mainstream artificial intelligence models with simple statistical methods across ten experimental datasets. The results showed that, on multiple datasets, traditional statistical methods performed comparably to or even better than complex AI models. This finding raises questions about existing model evaluation standards.
Maria Brbic, head of the MLBio laboratory, said: "The performance of simple methods is comparable to that of advanced AI models, which prompts us to think: do these complex models truly understand the effects of genetic changes? Are current evaluation metrics suitable for such models?"
To eliminate evaluation bias, the team developed the Systema tool. This tool effectively reduces interference caused by systematic differences in experiments and focuses on identifying the unique effects of gene perturbations. Ramon Viñas Torné, a postdoctoral researcher at MLBio and first author of the paper, pointed out: "Systema not only reduces the impact of systematic bias but also enhances the interpretability of the actual effects of gene perturbations."
Evaluation using Systema revealed that AI models still face challenges in predicting the effects of novel gene changes. Although some models can accurately predict gene changes belonging to the same biological process, overall prediction accuracy needs to be improved. The researchers recommend using biologically meaningful metrics to evaluate models, with a focus on the ability of predictions to explain cellular features.
The study suggests that in the future, larger and more diverse experimental datasets are needed to improve prediction accuracy. At the same time, new technologies capable of observing cell morphology and location should be integrated to deepen the understanding of the mechanisms underlying gene perturbation effects.