
en.Wedoany.com Reported - Researchers at Tohoku University have discovered a new catalyst design principle, revealing that dual-atom catalysts (DACs) exhibit a "dual-Sabatier optima" pattern in the oxygen reduction reaction, challenging the single-peak volcano model hypothesis that has been used for decades. This finding could pave the way for reducing the cost of hydrogen fuel cells.
Fuel cells are regarded as key devices for building a low-carbon society, generating electricity from hydrogen with clean emissions. However, many fuel cells still rely on precious metals such as platinum to drive the oxygen reduction reaction (ORR), a process that directly affects performance and cost. Traditional catalytic theory explains activity patterns using a "single-peak volcano" model, which posits that the best catalysts lie within a narrow range of chemical properties. However, when analyzing a large-scale experimental dataset from the Digital Catalysis Platform (DigCat), the research team found that dual-atom catalysts did not follow this expected pattern.
Using advanced theoretical simulations, microkinetic modeling, and machine learning, the researchers studied over 200 dual-atom catalysts. The results showed that DACs are primarily governed by a reaction pathway known as the dissociative mechanism, rather than the associative mechanism commonly observed in single-atom catalysts. This shift has a significant impact on catalyst activity: DACs no longer exhibit a single optimal performance peak but instead display two distinct optimal regions, i.e., "dual-Sabatier optima." The emergence of the two peaks stems from a shift in the rate-determining step during the reaction, switching between oxygen dissociation, oxygen protonation, and hydroxyl protonation.
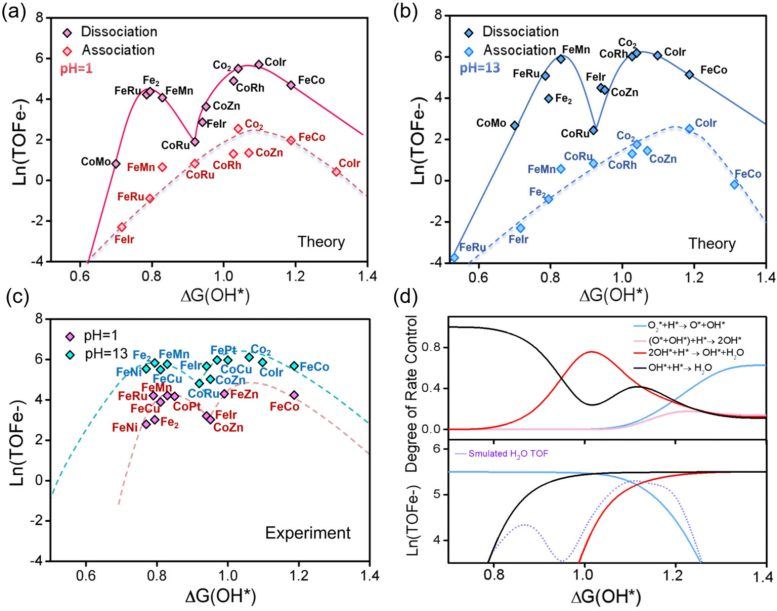
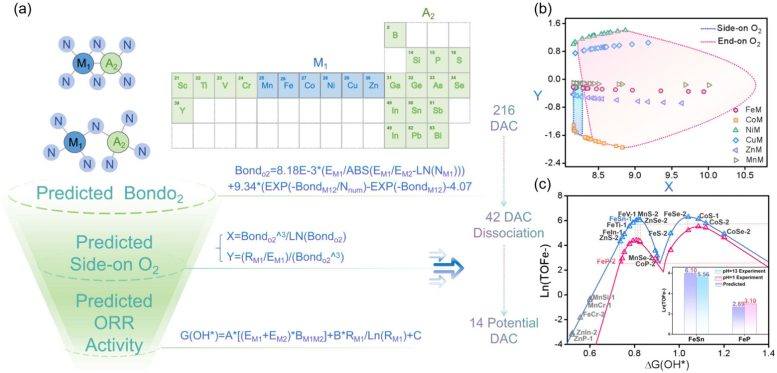
The researchers found that this principle applies to a variety of catalyst types, including systems composed of transition metals, metalloid elements, and even non-metal atoms. By combining interpretable machine learning with theoretical modeling, the team built a predictive framework capable of rapidly identifying promising catalyst structures. Hao Li, a distinguished professor at the Advanced Institute for Materials Research (WPI-AIMR) at Tohoku University, stated that the long-standing assumption has been that dual-atom catalysts follow the same activity rules as single-atom catalysts. However, the latest work shows that when two atoms work together, entirely different mechanisms may emerge, opening new opportunities for designing efficient materials for clean energy technologies.
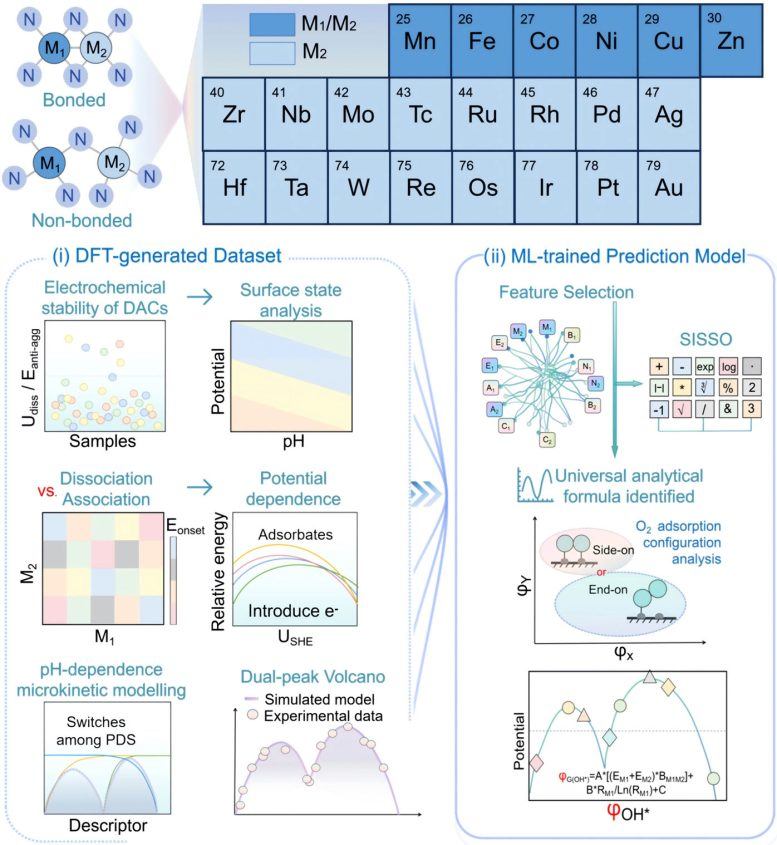
The potential impact of this discovery may extend beyond fuel cells, potentially guiding the development of catalysts for other energy conversion and chemical production processes. The research also demonstrates how artificial intelligence can extract hidden scientific patterns from existing experimental data, thereby accelerating the screening of new materials. Next, the team plans to apply this method to more complex multi-metal catalysts and other energy-related reactions beyond ORR. By integrating AI agents, machine learning, and electrochemical simulations into the DigCat platform, they aim to create a fully autonomous digital system for the rapid design of next-generation catalysts for sustainable energy.










